TroFuse-012 Studie
THERAPIESTUDIE ZUR PRÜFUNG VON SACITUZUMAB TIRUMOTECAN IN KOMBINATION MIT PEMBROLIZUMAB ALS WEITERFÜHRENDE BEHANDLUNG BEI PATIENT:INNEN MIT TRIPLE-NEGATIVEM BRUSTKREBS NACH EINER VORBEHANDLUNG VOR DER OPERATION
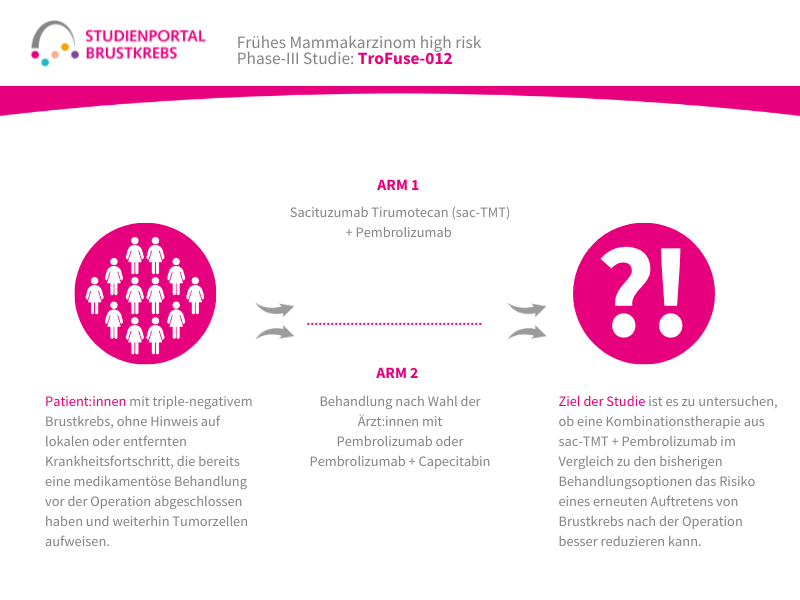
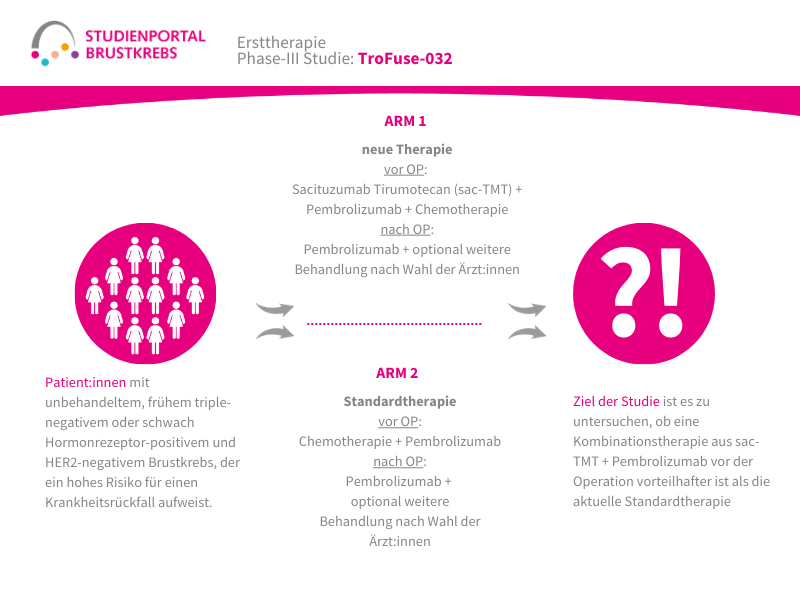
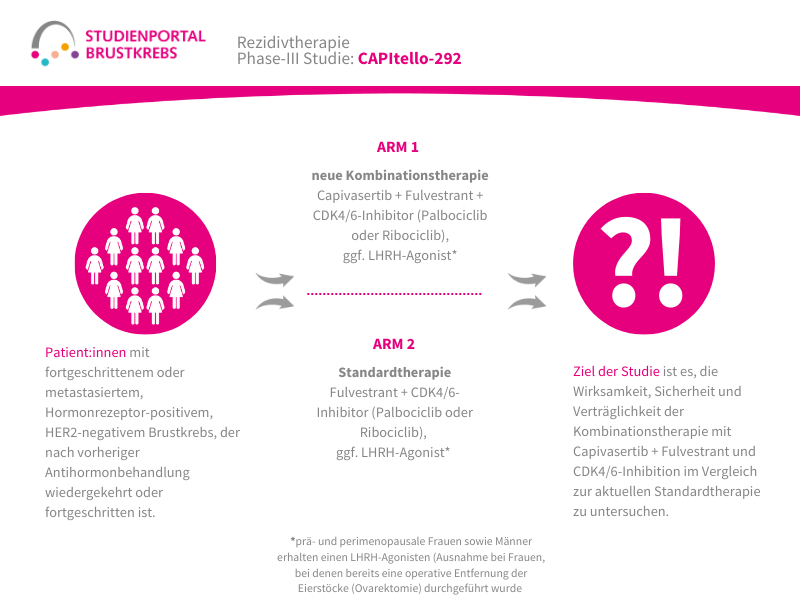
Die TroFuse-012-Studie ist eine randomisierte (die Zuordnung in die Behandlungsgruppe erfolgt nach dem Zufallsprinzip) Phase-3-Studie. Sie untersucht eine neue Behandlungsstrategie für Patient:innen mit triple-negativem Brustkrebs (TNBC), bei denen nach einer medikamentösen Behandlung vor der Operation noch Tumorzellen im entfernten Gewebe nachweisbar sind. Verglichen wird die Kombination aus den Studienmedikamenten Sacituzumab Tirumotecan (sac-TMT) und Pembrolizumab mit einer Behandlung mit Pembrolizumab allein oder mit Pembrolizumab in Kombination mit Capecitabin (die Therapie im Vergleichsarm erfolgt nach Wahl der behandelnden Ärzt:innen).
Was wird in dieser Studie untersucht?
Triple-negativer Brustkrebs (TNBC) ist eine besondere Form des Brustkrebses, bei der die Tumorzellen keine oder nur sehr geringe Mengen bestimmter Rezeptoren für Hormone (Östrogen und Progesteron) sowie des HER2-Proteins aufweisen. Dadurch können viele zielgerichtete oder hormonelle Therapien nicht eingesetzt werden. TNBC kann schneller wachsen und hat ein erhöhtes Risiko, nach der Behandlung erneut aufzutreten. Bei frühem, aber risikoreichem TNBC wird die Behandlung häufig bereits vor der Operation (neoadjuvante Therapie) begonnen. Dabei werden meist eine Chemotherapie und je nach Situation eine Immuntherapie eingesetzt. Wenn nach dieser Vorbehandlung bei der Operation noch lebende Tumorzellen im Brustgewebe oder in den Lymphknoten gefunden werden, besteht weiterhin ein erhöhtes Risiko für einen Rückfall.
Eine wichtige Rolle spielt dabei ein Protein namens TROP2, das auf der Oberfläche vieler triple-negativer Zellen besonders häufig vorkommt. Eine hohe TROP2-Menge wird mit einem aggressiveren Krankheitsverlauf in Verbindung gebracht. Der Wirkstoff sac-TMT ist eine moderne Antikörper-Wirkstoff-Verbindung. Es besteht aus einem Antikörper, der gezielt an TROP2 auf Krebszellen bindet und einem daran gekoppelten Zellgift, das direkt in die Krebszelle eingeschleust wird. Dadurch soll die Behandlung Tumorzellen gezielter angreifen.
Pembrolizumab ist eine Immuntherapie. Es blockiert ein Signal, mit dem Tumorzellen das Immunsystem bremsen können. Dadurch können körpereigene Abwehrzellen die Krebszellen besser erkennen und bekämpfen.
Was ist das Ziel der Studie?
Das Hauptziel der Studie ist zu untersuchen, ob die Kombination aus sac-TMT und Pembrolizumab im Vergleich zu den bisherigen Behandlungsoptionen das Risiko eines erneuten Auftretens von Brustkrebs nach der Operation besser reduzieren kann. Dabei wird insbesondere das invasive krankheitsfreie Überleben (iDFS) untersucht. Dieser Begriff beschreibt die Zeit nach der Operation, in der keine erneute invasive Brustkrebserkrankung, keine Metastasen und kein neuer invasiver Tumor auftritt. Zusätzlich wird untersucht, wie sicher und verträglich die Behandlung ist und ob sie das Gesamtüberleben der Patient:innen verlängern kann.
Wie ist der Ablauf der Studie?
Nach der Vorbehandlung vor der Operation und dem Nachweis, dass noch Tumorzellen vorhanden sind, werden die Patient:innen nach dem Zufallsprinzip einer von zwei Behandlungsgruppen zugeteilt:
Arm 1:
Sacituzumab Tirumotecan, intravenös alle zwei Wochen + Pembrolizumab intravenös alle 6 Wochen
Arm 2:
Behandlung nach Wahl der behandelnden Ärzt:innen (Pembrolizumab allein oder in Kombination mit Capecitabin)
Die Behandlung in beiden Studienarmen erfolgt für ca. 24 Wochen.
Während der Studie finden regelmäßige Untersuchungen statt, um den Gesundheitszustand, mögliche Nebenwirkungen und den weiteren Verlauf der Erkrankung zu überwachen.
Gibt es Risiken?
Über mögliche Risiken bzw. Nebenwirkungen, die mit der Teilnahme verbunden sind, werden Sie im Rahmen eines Aufklärungsgesprächs informiert.
Teilnahmevoraussetzungen
An dieser Studie können Männer und Frauen ab einem Alter von 18 Jahre teilnehmen, mit:
- Diagnose eines triple-negativem Brustkrebs
- bereits abgeschlossener medikamentöser Behandlung vor der Operation
- Kein Hinweis auf lokalen oder entfernten Krankheitsfortschritt
- nachweisbaren Tumorzellen im Brustgewebe oder in den Lymphknoten nach der Operation (keine vollständige Tumorentfernung durch die Vorbehandlung, keine pathologische Komplettremission)
Darüber hinaus gibt es aber auch noch weitere Kriterien, die für eine Teilnahme an der Studie erfüllt sein müssen. Interessierte Patient:innen sollten mit den Prüfärzt:innen an einem Studienzentrum sprechen, welche prüfen können, ob diese Studie für Sie in Frage kommt.
Wo kann ich an dieser Studie teilnehmen?
Weitere Informationen zu teilnehmende Zentren finden Sie hier:
https://studienportal-brustkrebs.de/deutschlandkarte
Diese Studie wird durchgeführt von: