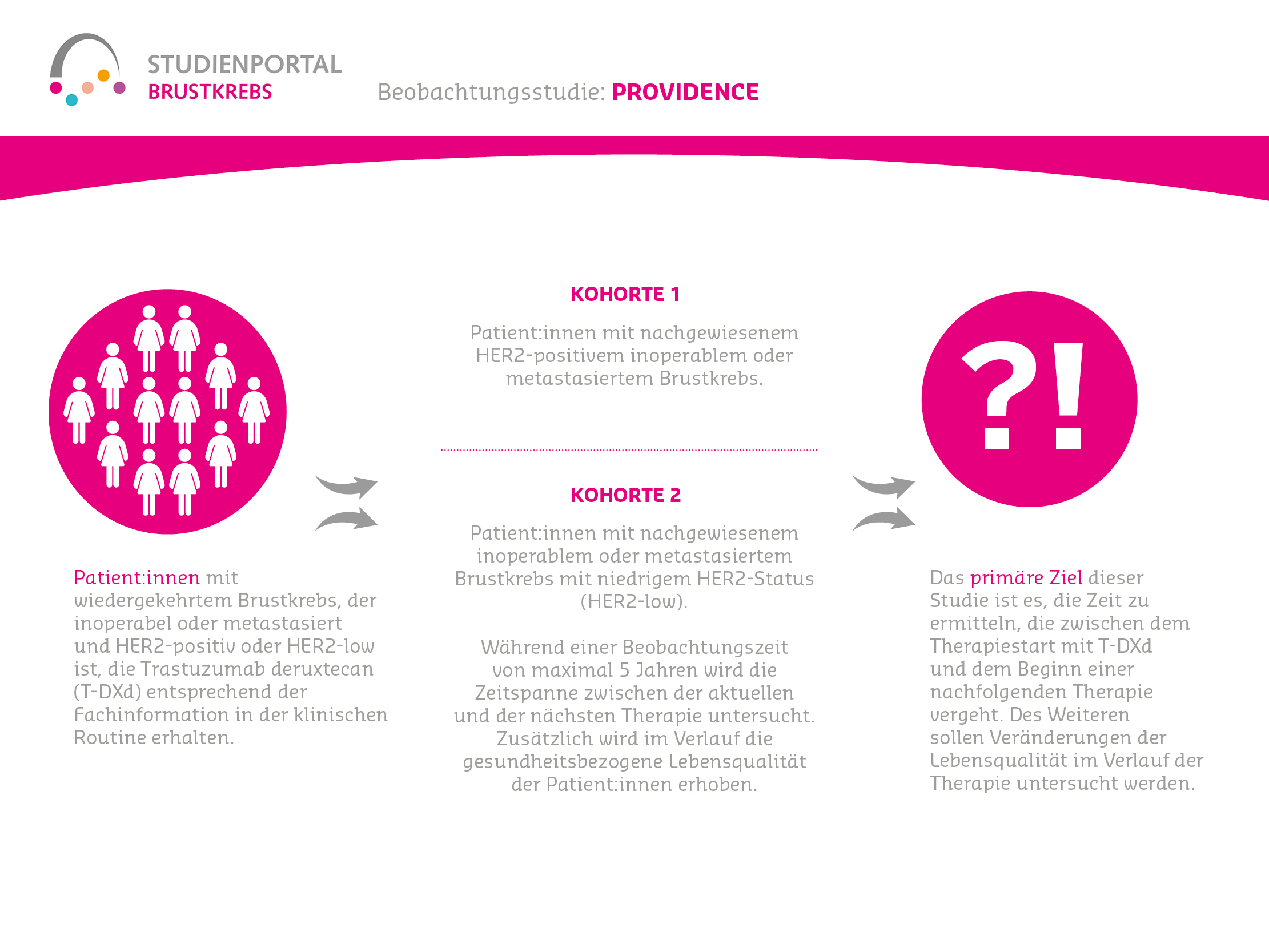
NOGGO B4 / VICTORIA-Studie
BEOBACHTUNGSSTUDIE ZUR WIRKSAMKEIT UND LEBENSQUALITÄT UNTER TRODELVY® BEI PATIENT:INNEN MIT FORTGESCHRITTENEM ODER NICHT OPERIERBAREM HER2-NEGATIVEM BRUSTKREBS
Die VICTORIA-Studie ist eine Beobachtungsstudie. Das bedeutet, dass die Patient:innen eine Behandlung (für diese Studie mit Trodelvy® (Sacituzumab Govitecan)) im Rahmen der normalen Versorgung erhalten und die Studienärzt:innen lediglich Daten rund um die Behandlung erfassen. Ziel dabei ist es, zu untersuchen, wie wirksam und verträglich die Therapie im klinischen Alltag in Deutschland ist und wie sich die Behandlung auf die Lebensqualität auswirkt.
Was wird in dieser Studie untersucht?
Bestimmte Untergruppen von HER2-negativem Brustkrebs sind besonders schwer zu behandeln:
- Dreifach negativer Brustkrebs (TNBC): Diese Tumoren haben keine Hormonrezeptoren (ER-/PR-) und kein HER2-Protein auf ihrer Zelloberfläche und bieten somit weniger Angriffspunkte für zielgerichtete Medikamente. Sie sind meist aggressiver, haben eine schlechtere Prognose und führen häufig zu belastenden Behandlungsverläufen.
- Hormonrezeptor-positiver, HER2-negativer Brustkrebs (HR+/HER2-): Diese Form tritt häufiger auf. Die Prognose ist etwas günstiger, dennoch stoßen die Therapien im fortgeschrittenen Stadium oft an Grenzen, da die Tumoren resistent gegen Medikamente werden können.
Für beide Gruppen sind die Behandlungsmöglichkeiten eingeschränkt, da zielgerichtete HER2-Therapien nicht greifen. Beim TNBC kommt erschwerend hinzu, dass meist keine Hormontherapien wirken. Daher wird bisher oft eine Chemotherapie eingesetzt.
Sacituzumab Govitecan ist ein sogenanntes Antikörper-Wirkstoff-Konjugat:
Eine Verbindung aus einem Antikörper, der gezielt eine Struktur (z. B. ein Protein oder Rezeptor) auf der Oberfläche der Krebszelle erkennt und einem Medikament. In diesem Fall ist an diesen Antikörper ein Chemotherapie-Wirkstoff gekoppelt, der direkt in die Tumorzelle eingeschleust wird. So können die Krebszellen gezielt zerstört werden, während gesundes Gewebe besser geschont werden kann.
In klinischen Studien konnte Sacituzumab Govitecan das Überleben verlängern und die Lebensqualität verbessern, sowohl bei Patient:innen mit TNBC als auch mit HR+/HER2- Brustkrebs. Nun soll überprüft werden, dass sich diese Ergebnisse auch in der Anwendung im klinischen Alltag bestätigen lassen.
Was ist das Ziel der Studie?
Das Hauptziel ist es, das Gesamtüberleben von Patient:innen mit fortgeschrittenem TNBC oder HR+/HER2- Brustkrebs unter Therapie mit Sacituzumab Govitecan in der klinischen Praxis zu beschreiben. Darüber hinaus untersucht die Studie noch die Zeit ohne das Voranschreiten der Erkrankung, die Sicherheit und Verträglichkeit des Medikaments und die Lebensqualität der Patient:innen unter Therapie.
Wie ist der Ablauf der Studie?
In dieser Beobachtungsstudie werden ausschließlich Daten der klinischen Routine gesammelt. Die Teilnahme an der Studie hat keinen Einfluss auf die Behandlung der Patient:innen. Diese erfolgt unabhängig von der Studienteilnahme gemäß ärztlicher Praxis und geltender Leitlinien. Zusätzliche Besuche in der Klinik/ Praxis sind nur erforderlich für das Ausfüllen der Fragebögen zur Lebensqualität, wenn die Therapie frühzeitig abgebrochen wird. Nach dem schriftlichen Einverständnis zur Teilnahme an der VICTORIA-Studie wird mit der Dokumentation von Daten begonnen.
Zu Beginn werden zum Beispiel demografische Daten der Patient:innen und die Krankengeschichte der letzten 24 Monate, sowie Fragebögen zur aktuellen Lebensqualität erfasst. Die Fragebögen zur Lebensqualität werden ab da in regelmäßigen Abständen erneut ausgefüllt (3, 6 und 12 Monate nach Therapiebeginn). Nach abgeschlossener Therapie oder sollte die Erkrankung während der Behandlung fortschreiten, dann gibt es 90 Tage nach letzter Therapiegabe noch eine letzte Befragung zur Lebensqualität und eine abschließende Dokumentation von Nebenwirkungen.
Gibt es Risiken?
Über mögliche Risiken, die mit der Teilnahme verbunden sind, werden Sie im Rahmen eines Aufklärungsgesprächs informiert. Da es sich bei dieser Beobachtungsstudie um eine reine Dokumentation der klinischen Routine ohne geplante zusätzliche Behandlung handelt, ist mit der Teilnahme kein zusätzliches gesundheitliches Risiko verbunden.
Teilnahmevoraussetzungen
An dieser Studie können Frauen und Männer ab einem Alter von 18 Jahren teilnehmen, mit:
- inoperablem und/oder metastasiertem Brustkrebs, welcher:
- dreifach negativ (HR- und HER2-) ist oder
- Hormonrezeptor-positiv und HER2-negativ ist.
- Eine Behandlung nach aktuellem Standard mit Sacituzumab Govitecan ist geplant auf Grund der Tumorbeschaffenheit und bereits vorangegangener Therapien.
Darüber hinaus gibt es aber auch noch weitere Kriterien, die für eine Teilnahme an der Studie erfüllt sein müssen. Interessierte Patient:innen sollten mit den Prüfärzt:innen an einem Studienzentrum sprechen, welche prüfen können, ob diese Studie für sie in Frage kommt.
Wo kann ich an dieser Studie teilnehmen?
Weitere Informationen zu teilnehmende Zentren finden Sie hier:
https://studienportal-brustkrebs.de/deutschlandkarte
Diese Studie wird durchgeführt von: