HERTHENA-Breast04
THERAPIESTUDIE ZUR PRÜFUNG VON PATRITUMAB DERUXTECAN BEI PATIENT:INNEN MIT HORMONREZEPTOR-POSITIVEM, HER2-NEGATIVEM FORTGESCHRITTENEM BRUSTKREBS
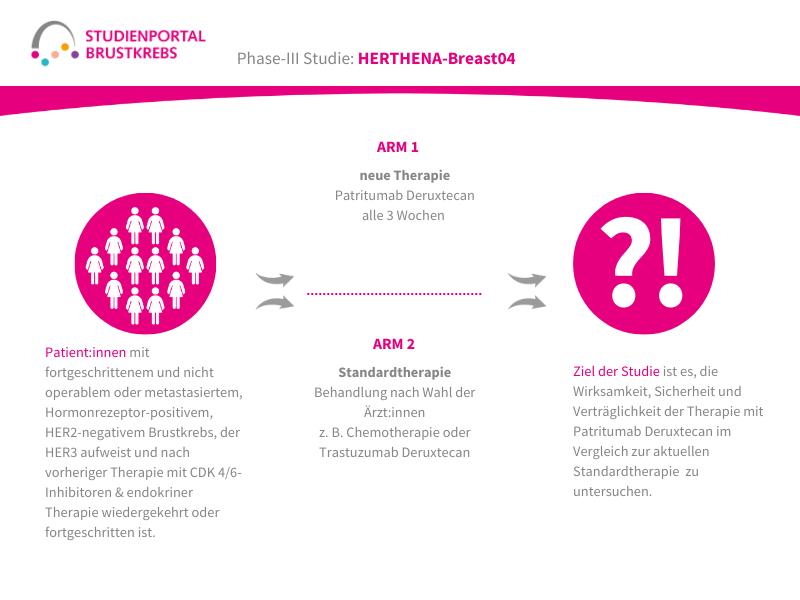
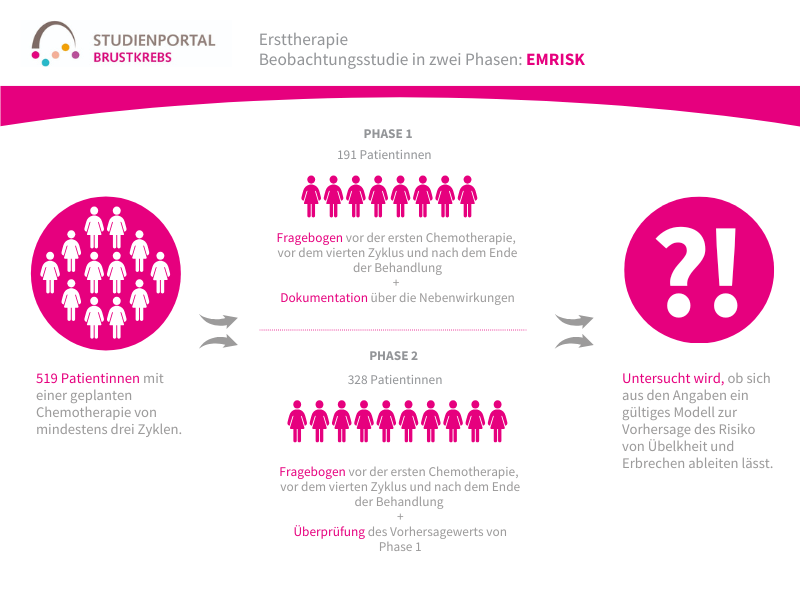
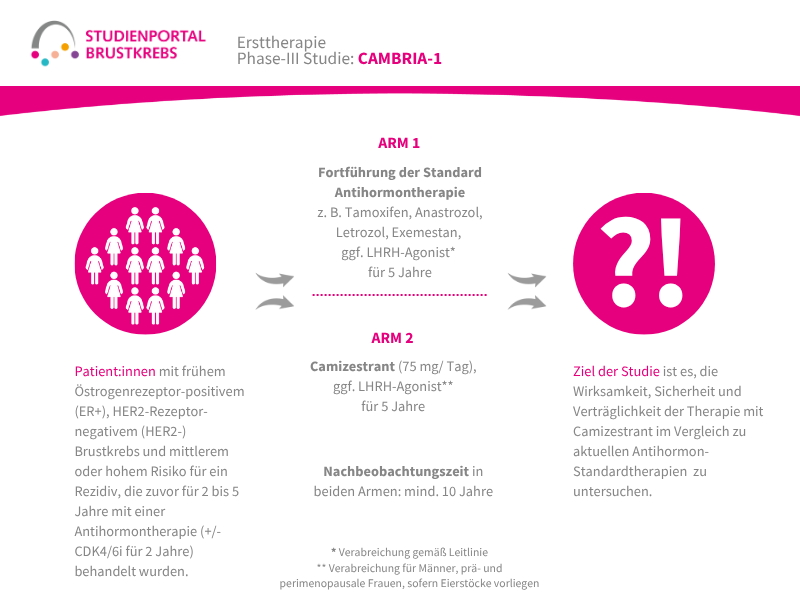
Die HERTHENA-Breast04-Studie ist eine internationale, randomisierte (die Zuordnung in die Behandlungsgruppe erfolgt nach dem Zufallsprinzip) Phase-3-Studie. In ihr wird ein neues Medikament namens Patritumab Deruxtecan mit den aktuellen Standardbehandlungen verglichen. Die Studie richtet sich an Patient:innen mit Hormonrezeptor-positivem (HR-positiv), HER2-negativem Brustkrebs, der lokal fortgeschritten und nicht mehr vollständig operierbar oder bereits metastasiert ist. Ziel ist es herauszufinden, ob Patritumab Deruxtecan das Fortschreiten der Erkrankung verlangsamen und/oder das Leben verlängern kann.
Was wird in dieser Studie untersucht?
Nicht mehr operierbarer oder bereits metastasierter Brustkrebs wird gemäß aktuellem Stand überwiegend mit einer endokrinen Behandlung (Hormontherapie) therapiert, häufig kombiniert mit weiteren Medikamenten. Wenn diese Behandlung nicht mehr ausreichend wirkt, kommen meist Chemotherapien zum Einsatz. Trotzdem bleibt die Erkrankung schwer behandelbar.
Patritumab Deruxtecan ist ein sogenanntes Antikörper-Wirkstoff-Konjugat (ADC).
Es besteht aus zwei Teilen: einem Antikörper, der gezielt an HER3, ein Protein auf der Oberfläche vieler Brustkrebszellen, bindet und einem daran gekoppelten Krebsmedikament (Deruxtecan), das direkt in die Tumorzelle eingeschleust wird. In der Zelle bricht die Verbindung und Deruxtecan wird freigesetzt, um die Tumorzelle zu schädigen.
HER3 (humaner epidermaler Wachstumsfaktor-Rezeptor 3) ist ein Rezeptor auf der Oberfläche von Zellen, das Signale für Wachstum und Überleben weitergeben kann. Besonders Krebszellen von HR+/HER2– Tumoren bilden vermehrt HER3, wodurch sie widerstandsfähiger gegen Behandlungen werden können. Da HER3 vor allem auf Tumorzellen und weniger auf gesunden Zellen vorkommt, eignet es sich gut als zielgerichteter Angriffspunkt für moderne Therapien wie Patritumab Deruxtecan, während gesundes Gewebe möglichst geschont wird.
Verglichen wird dieser neue Therapieansatz mit einer Standardbehandlung, die von den behandelnden Ärzt:innen entschieden wird. Das bedeutet, dass Patient:innen in der Kontrollgruppe kein fest vorgegebenes Medikament, sondern eine aus mehreren zugelassenen und in den Leitlinien empfohlenen Standardtherapien, die für die/den Patientin/Patienten am besten geeignet ist, bekommen.
Was ist das Ziel der Studie?
Hauptziel der HERTHENA-Breast04 Studie ist es zu untersuchen, ob eine Behandlung mit Patritumab Deruxtecan einen Vorteil bringt, indem die Behandlung das Fortschreiten der Erkrankung aufhält und die Überlebenszeit verlängert. Daneben wird auch die Sicherheit und die Lebensqualität während der Therapie geprüft.
Wie ist der Ablauf der Studie?
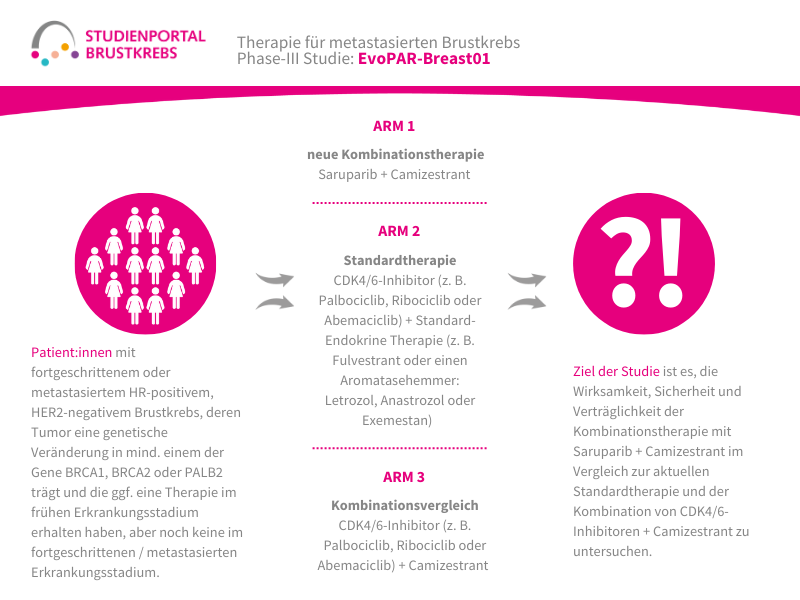
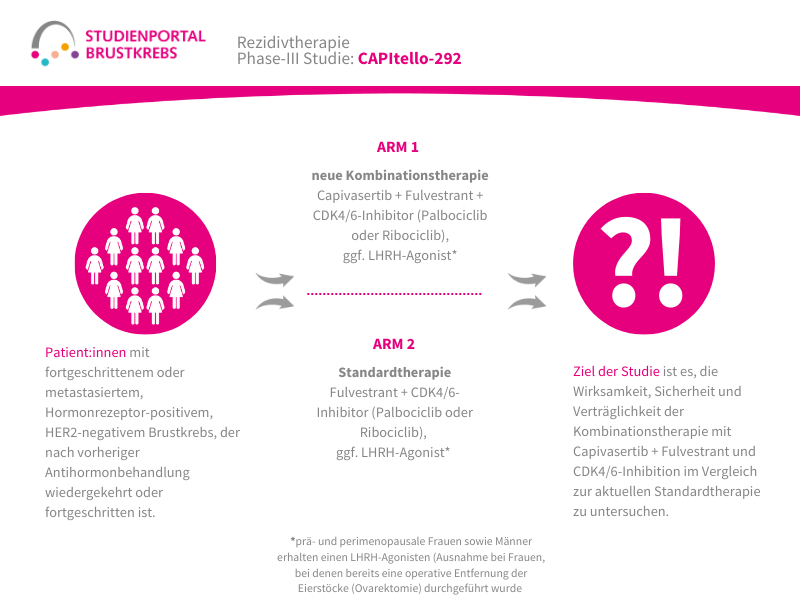
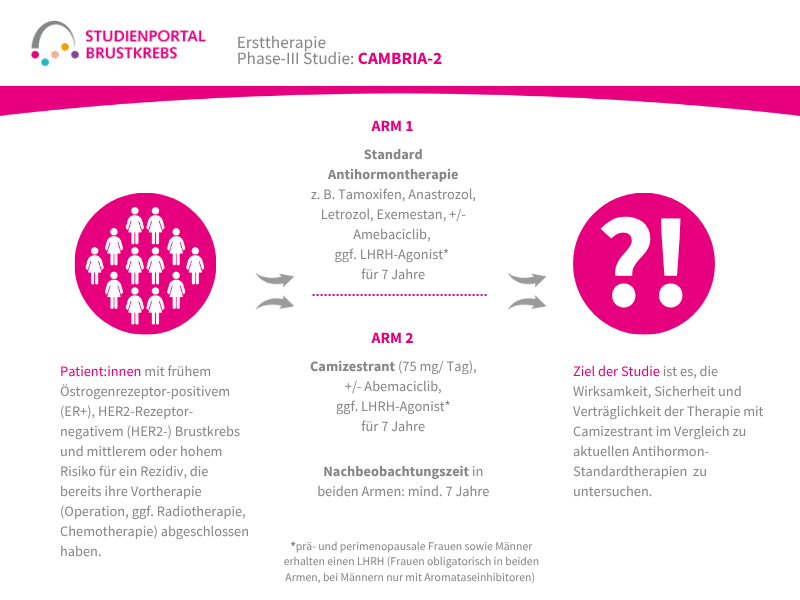
Im Rahmen der Studie gibt es zwei Behandlungsarme, denen die Patient:innen in einem Verhältnis von 1:1 zufällig zugeordnet werden:
Arm 1:
Patritumab Deruxtecan – Infusion alle 3 Wochen
Arm 2: (Kontrollgruppe)
Behandlung nach Wahl der Ärzt:innen – z. B. Paclitaxel; Nab-Paclitaxel; Capecitabin; Liposomales Doxorubicin oder Trastuzumab Deruxtecan
Die Behandlung in beiden Armen erfolgt bis zum Fortschreiten der Erkrankung, danach werden die Patient:innen für maximal 7 Jahre nachbeobachtet.
Gibt es Risiken?
Über mögliche Risiken bzw. Nebenwirkungen, die mit der Teilnahme verbunden sind, werden Sie im Rahmen eines Aufklärungsgesprächs informiert.
Teilnahmevoraussetzungen
An dieser Studie können Männer und Frauen ab einem Alter von 18 Jahre teilnehmen, mit:
- Diagnose eines Hormonrezeptor-positiven / HER2-negativen Brustkrebs, der entweder lokal fortgeschritten und nicht operabel oder metastasiert ist
- Tumor weist HER3 auf aus einer Biopsie, die an einer entfernten Metastasenstelle oder vom lokal fortgeschrittenen Tumor während oder nach der letzten Therapie durchgeführt wurde
- Nachgewiesenem Fortschritt oder Rückfall der Erkrankung unter einer vorherigen Therapie mit Cyclin-abhängigen Kinase (CDK)4/6-Inhibitoren + endokriner Therapie
- Messbarer Resterkrankung
Darüber hinaus gibt es aber auch noch weitere Kriterien, die für eine Teilnahme an der Studie erfüllt sein müssen. Interessierte Patient:innen sollten mit den Prüfärzt:innen an einem Studienzentrum sprechen, welche prüfen können, ob diese Studie für Sie in Frage kommt.
Wo kann ich an dieser Studie teilnehmen?
Weitere Informationen zu teilnehmende Zentren finden Sie sehr bald hier:
https://studienportal-brustkrebs.de/deutschlandkarte
Diese Studie wird unterstützt von: